解决AlphaFold 2训练数据私有难题:哈佛、哥大开源1600万组蛋白质序列!
来源:51CTO.COM
时间:2023-09-20 18:25:12 238浏览 收藏
在科技周边实战开发的过程中,我们经常会遇到一些这样那样的问题,然后要卡好半天,等问题解决了才发现原来一些细节知识点还是没有掌握好。今天golang学习网就整理分享《解决AlphaFold 2训练数据私有难题:哈佛、哥大开源1600万组蛋白质序列!》,聊聊,希望可以帮助到正在努力赚钱的你。
蛋白质是生命的主力军,了解它们的序列和结构,是设计新酶、开发救命药物等生物学和医学挑战的关键。
AlphaFold 2是DeepMind开发的一种具有前所未有准确性的蛋白质结构预测工具
然而,因为缺乏可供开放训练的数据,这个领域的发展受到了严重的阻碍
来自哈佛大学、哈佛医学院、哥伦比亚大学、纽约大学和Flatiron Institute的研究人员,引入了一个开源数据库
这个名为OpenProteinSet的开源数据库,可以通过大规模提供蛋白质比对数据,来大大改善这种状况。
提供的数据集与用于训练AlphaFold 2的数据集具有相同的质量
因为AlphaFold 2,MSA的实用性爆炸性增长
蛋白质的功能是通过氨基酸序列进行编码的
在进化过程中,这些序列会积累一些微小的变化,而蛋白质的整体结构和功能却一直保持不变。
多序列对齐(MSA)是一种与进化相关的蛋白质序列组,通过插入间隙来进行对齐,以便将匹配的氨基酸最终放置在同一列中
通过对这些MSA中的模式进行分析,我们可以更深入地了解蛋白质的结构和功能
MSA的每一行都代表一个蛋白质序列。蛋白质由20个氨基酸(或称为“残基”)组成,形成一维字符串,每个氨基酸或“残基”用一个字母表示
在MSA的第一行中,我们给出了目标或「查询」蛋白质。接下来的行是根据与查询序列的相似性,从大型序列数据库中检索到的进化相关(「同源」)蛋白质
为了改进比对、适应长度随时间变化的同源序列,MSA比对软件可以在同源序列中插入「缺口」(此处用破折号表示)或删除残基。
MSA中同源序列的数量(即"深度")和多样性对于MSA的实用性都是有益的
 MSA引物
MSA引物
在过去的许多年里,蛋白质的研究一直对多序列比对(MSA)至关重要。然而,随着AlphaFold 2的问世,2021年的实用性呈现了爆炸性的增长
通过MSA,AlphaFold 2能够以近乎实验级的准确性预测蛋白质结构。
AlphaFold 2是开源的,但是仍然存在一个问题:它的训练数据是私有的
根据目标序列长度和正在搜索的序列数据库的大小,生成一个具有高灵敏度的MSA可能需要几个小时,这样做的计算成本很高
这样,蛋白质机器学习和生物信息学的前沿研究除了少数大型研究团队外,其他所有人都无法访问。
1600万个MSA全部开源
因此,团队提出了OpenProteinSet,这是一个在AlphaFold 2及其以上规模训练生物信息学的模型。
它包含了AlphaFold 2未发布的训练集,包括所有唯一的蛋白质数据库(PDB)链的MSAs和结构模板。
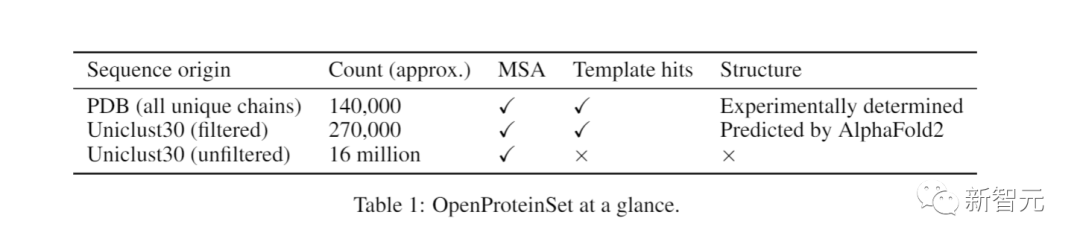
目前,OpenProteinSet已经提供了1600万个多序列比对(MSA)和相关数据,并且全部开源
PDB是确定蛋白质结构的权威数据库,而OpenProteinSet包含了PDB中所有140,000种蛋白质的多序列比对(MSA)
它还包括了来自UniProt知识库的序列,这些序列是按照相似性进行聚类的
OpenProteinSet可以为PDB蛋白质提供来自多个序列数据库的原始MSA
通过搜索PDB,它还能找到结构相似的蛋白质。
AlphaFold 2预测的结构涵盖了270,000个不同的UniProt集群
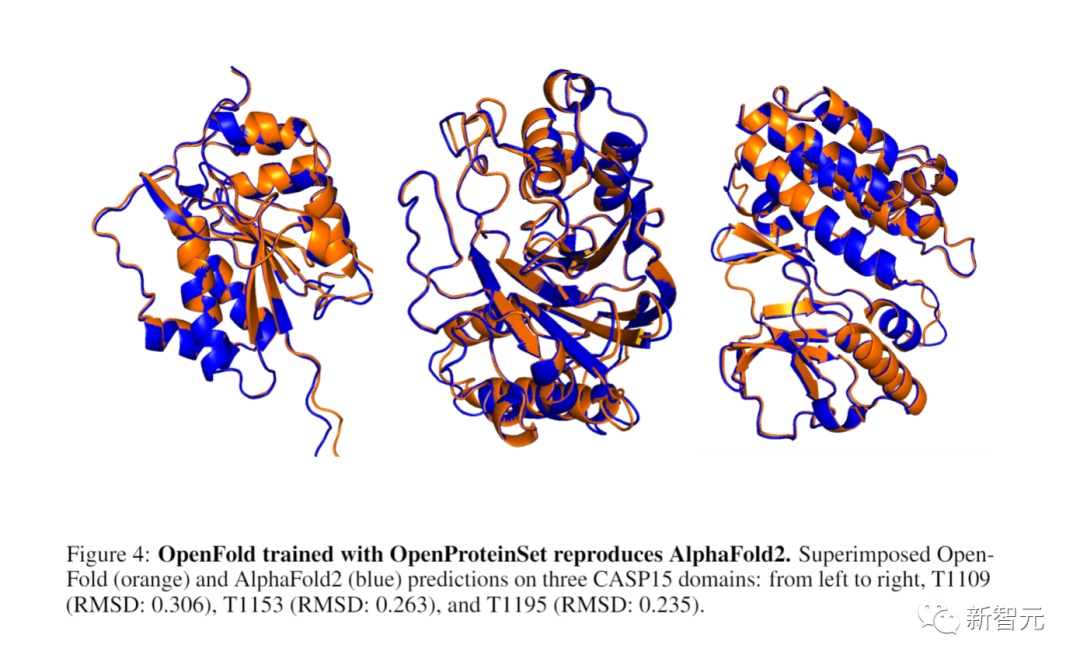
使用开源数据集重新创建AlphaFold 2
开发者还会使用OpenProteinSet来训练OpenFold,这是AlphaFold 2的一个开放版本。
他们发现OpenFold的性能与DeepMind的原始数据相当,证明了开放数据的充分性
团队表示,他们通过OpenProteinSet显著增加了分子机器学习社区可用的预计算MSA的数量和质量
这个数据集可以直接用于各种结构生物学任务
实验方法
OpenProteinSet是由超过1600万个独特的多序列比对(MSA)组成的,这些MSA是根据AlphaFold2论文中的算法生成的
这一计数包括截至2022年4月PDB中所有14万个唯一链的MSAs,以及针对同一数据库为Uniclust30中的每个序列集群计算的1,600万个MSAs。
在后续的研究中,研究人员确定了270,000个具有最大多样性的代表性集群,例如可以用于AphaFold2训练过程中的自我蒸馏集合
对于每个PDB链,研究者使用了不同的对齐工具和序列数据库计算三个MSAs。
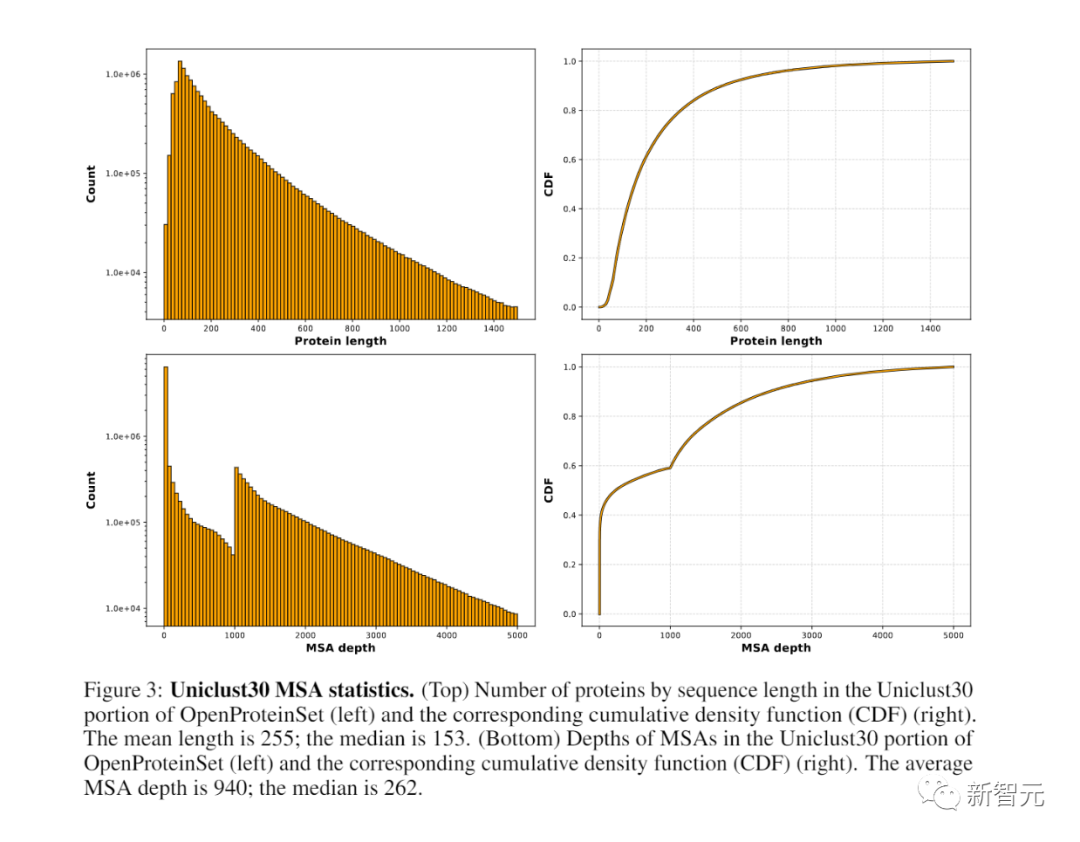
使用OpenFold中的脚本,可以从公开可用的PDBmmCIF文件中,检索相应的结构。
与生成AIphaFold2训练集的过程相同,研究人员对MSA生成工具的一些默认选项进行了修改
在接下来的过程中,大约产生了1600万个MSAs,每个集群都有一个
为了创建一个不同的、深度的MSAs子集,研究者通过迭代去除代表性链出现在其他MSAs中最多的MSAs。
不需要改变原本的意思,需要重写的语言是中文。将其重写为:这样重复,直到每个代表链只出现在它自己的MSA中
为了与对应的(未发布的)AlphaFold 2集进行奇偶性检验,研究者进一步删除了代表序列大于1024个残基或小于200个残基的簇。
最后,他们剔除了相应MSAs少于200个序列的簇,只剩下270,262个MSAs。
总的来说,OpenProteinSet中的MSAs代表了超过400万小时的计算。
OpenProteinSet大大提高了分子机器学习社区可用的预计算MSAs的数量和质量,它可以直接应用于结构生物学中的各种任务。
随着模型对数据的需求越来越大,像OpenProteimnSet这样的数据库既可以作为多模态语言模型的生物知识宝库,也可以作为多模态训练本身的实证研究工具。
总结一下,OpenProteinSet将在生物信息学、蛋白质机器学习等领域推动研究的进一步发展
以上就是本文的全部内容了,是否有顺利帮助你解决问题?若是能给你带来学习上的帮助,请大家多多支持golang学习网!更多关于科技周边的相关知识,也可关注golang学习网公众号。
-
501 收藏
-
501 收藏
-
501 收藏
-
501 收藏
-
501 收藏
-
191 收藏
-
299 收藏
-
科技周边 · 人工智能 | 6天前 | 人工智能 · mcp · ai agent · 工具接入 · 安全审计 · AI Agent MCP Model Context Protocol 工具清单 资源上下文 权限审计378 收藏
-
195 收藏
-
453 收藏
-
202 收藏
-
419 收藏
-
170 收藏
-
475 收藏
-
科技周边 · 人工智能 | 1星期前 | 人工智能 · tracing · ai agent · 可观测性 · 工具调用 · 可观测性 AI Agent Tracing 工具调用 OpenAI Agents SDK292 收藏
-
379 收藏
-
394 收藏
-

- 前端进阶之JavaScript设计模式
- 设计模式是开发人员在软件开发过程中面临一般问题时的解决方案,代表了最佳的实践。本课程的主打内容包括JS常见设计模式以及具体应用场景,打造一站式知识长龙服务,适合有JS基础的同学学习。
- 立即学习 543次学习
-

- GO语言核心编程课程
- 本课程采用真实案例,全面具体可落地,从理论到实践,一步一步将GO核心编程技术、编程思想、底层实现融会贯通,使学习者贴近时代脉搏,做IT互联网时代的弄潮儿。
- 立即学习 516次学习
-

- 简单聊聊mysql8与网络通信
- 如有问题加微信:Le-studyg;在课程中,我们将首先介绍MySQL8的新特性,包括性能优化、安全增强、新数据类型等,帮助学生快速熟悉MySQL8的最新功能。接着,我们将深入解析MySQL的网络通信机制,包括协议、连接管理、数据传输等,让
- 立即学习 500次学习
-

- JavaScript正则表达式基础与实战
- 在任何一门编程语言中,正则表达式,都是一项重要的知识,它提供了高效的字符串匹配与捕获机制,可以极大的简化程序设计。
- 立即学习 487次学习
-

- 从零制作响应式网站—Grid布局
- 本系列教程将展示从零制作一个假想的网络科技公司官网,分为导航,轮播,关于我们,成功案例,服务流程,团队介绍,数据部分,公司动态,底部信息等内容区块。网站整体采用CSSGrid布局,支持响应式,有流畅过渡和展现动画。
- 立即学习 485次学习